Harmful RNAs as stress signals
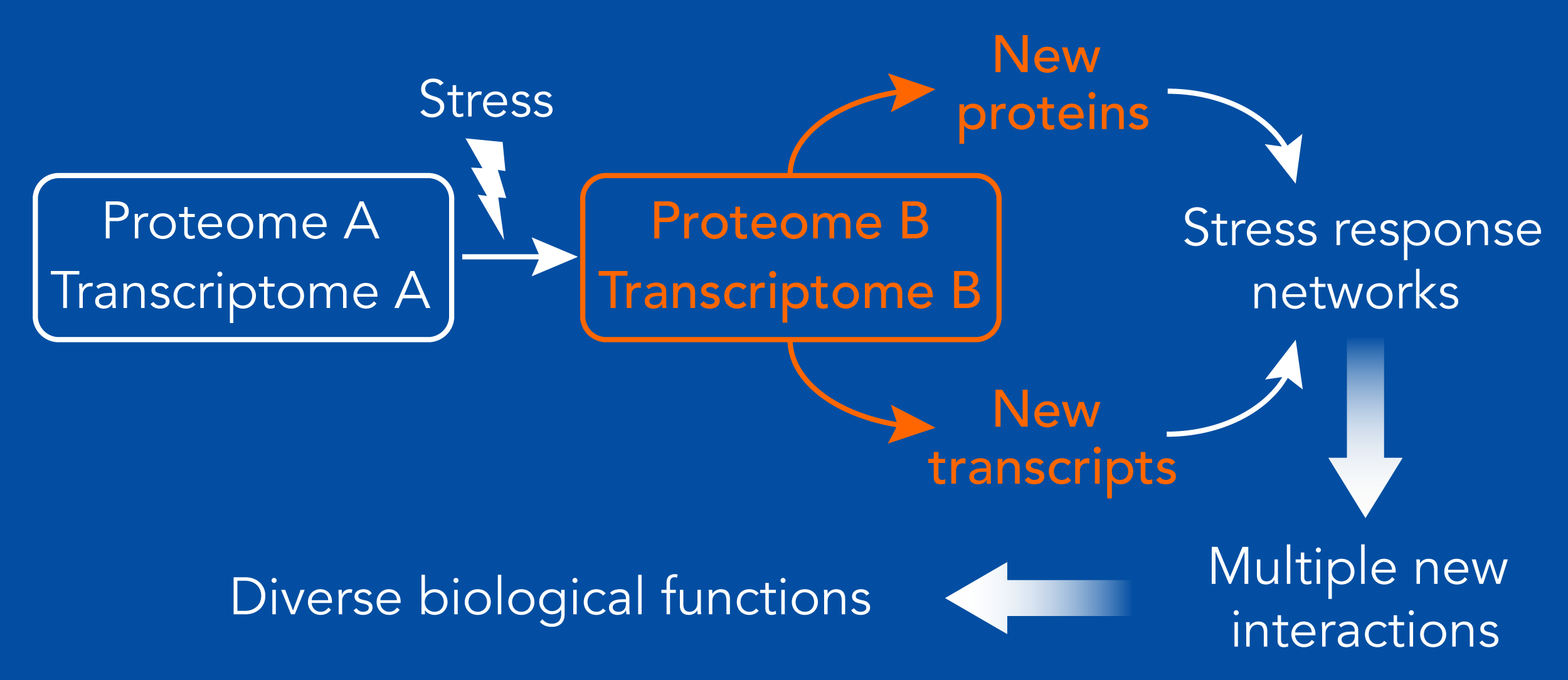
Activation of cellular stress responses leads to global changes in the proteome and transcriptome which in turn seed new biological interactions between multiple RNAs and proteins. These interactions can lead to different biological outcomes. Multiplying the interactions between several molecules can promote network-level rearrangements that allow adaptation in cells that face challenging conditions. We have recently shown that such network-level rearrangements occur in cancer cells in which proteasome function was compromised (Acosta-Alvear, et al., eLife 2015). Similarly, we anticipate that different interactions between RNAs and proteins can lead to different biological functions.
To understand how the RNA stress response (RSR) network is wired, we must first understand the input signals: the harmful RNAs that initiate it. Towards that end, we are developing a deep-sequencing based approach to identify harmful RNAs in living cells. This new technique, allows us to determine the defining features -sequence and structure- of harmful RNAs. Together with cross-linking and immunoprecipitation (CLIP)-based methods, which allow the identification of direct interactions between specialized RNA-binding proteins and toxic RNAs and in living cells in a transcriptome-wide manner, our methods allow defining the rules of RNA toxicity in living cells. Through computational and biochemical approaches, we study the structure-sequence determinants that make up "toxic RNA stress signatures”. The structures RNAs adopt can tailor how they interact with proteins, making the RNA an active participant in the “decision making” process once a protein-RNA interaction is established. We have recently illustrated this concept in the discovery of an RNA-embedded molecular “zipper” in the transcript encoding the XBP1 transcription factor. This RNA feature is instrumental for the IRE1-mediated splicing reaction that is central to the endoplasmic reticulum stress response (Peschek, Acosta-Alvear, et al., EMBO Reports 2015).
While the methods mentioned above are powerful for identifying the input signals for RSR and the RNA-protein interactions within it, additional layers of regulation are expected to arise from protein-protein interactions and post-translational modifications occurring within the network. These interactions have the potential to calibrate the RNA stress sensors. These additional layers of control can be identified by mass spectrometry after pulling down on key RSR components. By coupling the findings from all of these approaches we expect to discover the basic weaving of the network that allows the cell to tailor exquisite responses to distinguish healthy from harmful RNAs.




