The viral pirates: Hijacking proteostasis
Viruses must ensure that their peptides are folded into functional proteins. What are the tactics that enable viruses to hijack the protein folding and quality control machinery of the cell?
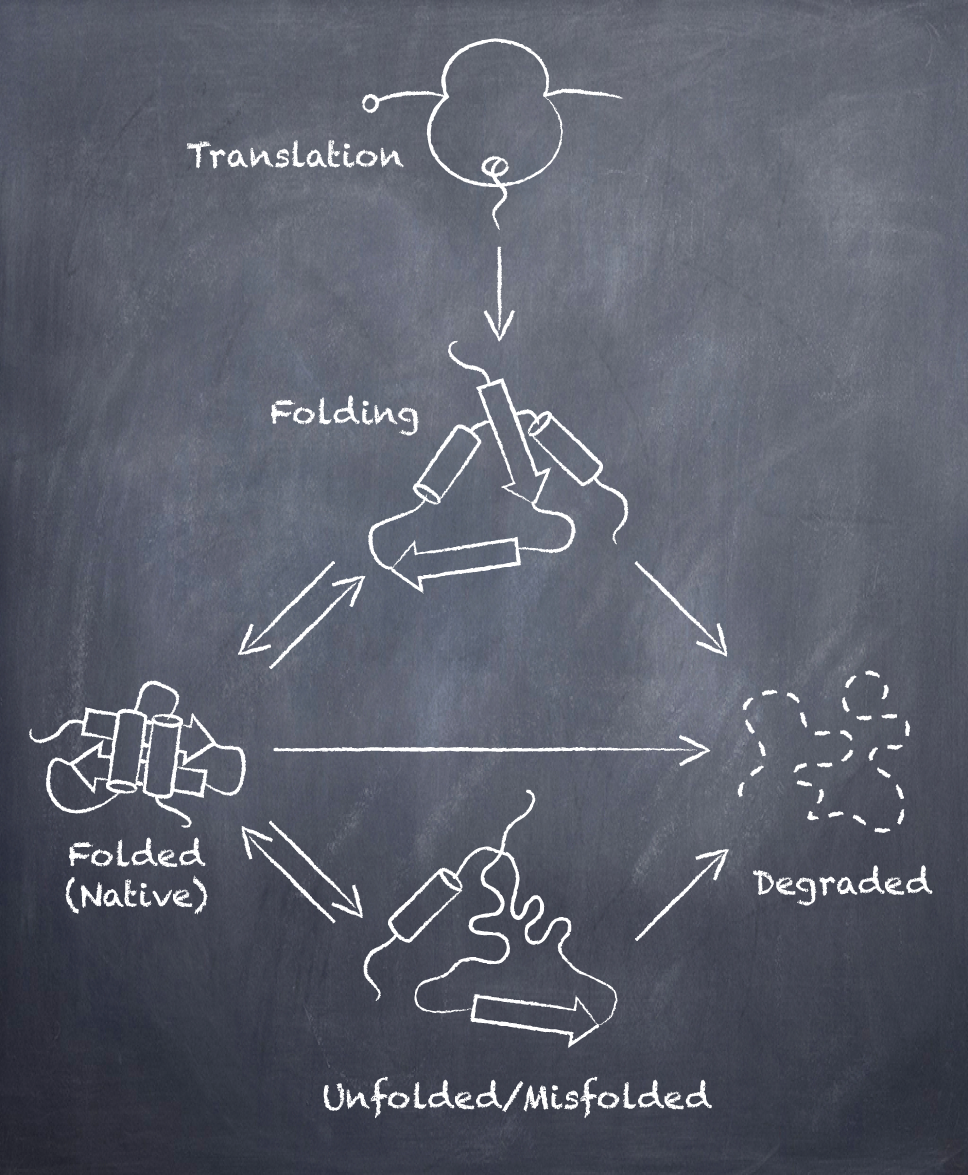
Cells maintain a healthy proteome by tightly regulating the synthesis, folding and degradation of proteins. The collection of regulatory pathways in charge of controlling these processes is known as the proteostasis network. Viruses interface with this network at multiple levels ensuring that the newly made viral proteins are correctly folded and modified during infection. Similar to their strict dependence on the translation initiation complex, viruses also rely on the host molecular chaperones and protein quality control systems for the maturation of their proteins. Our group is particularly interested in unraveling how viruses manipulate protein synthesis, folding and turnover in the endoplasmic reticulum (ER).
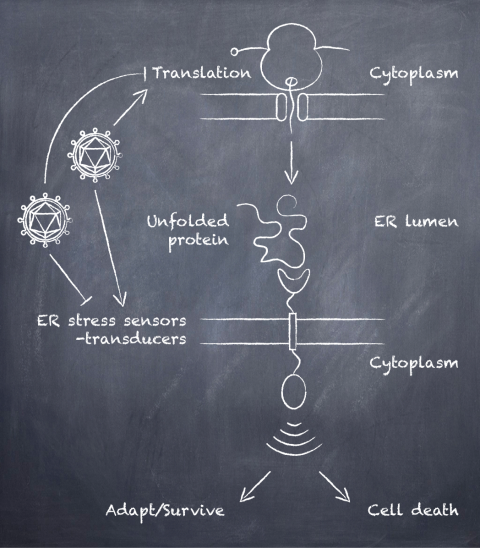
During infection, an increased influx of viral client proteins into the ER can overwhelm the normal function of the organelle. This unanticipated protein folding burden can lead to ER stress, which if not mitigated, can result in cell death. The unfolded protein response is an evolutionarily conserved regulatory network that oversees proper ER function and responds to cellular stress. In mammals, the UPR consists of three interconnected signaling pathways governed by ER resident transmembrane sensors/transducers that detect protein folding perturbations and initiate appropriate responses, namely the kinases IRE1 and PERK, and the membrane-tethered transcription factor ATF6. The concerted actions of IRE1, PERK, and ATF6 prevent further protein loading into the ER, expand the protein folding and degradative capacities of the ER, and if the stress cannot be resolved, promote cell death. Viruses modulate the activities of these sensor/transducers and their downstream effectors to facilitate and promote the folding of their own proteins while preventing premature cell death. Previous work by us and others has shown that herpesviruses manipulate the UPR (1). In ongoing work, we have evidence that KSHV impairs signaling downstream of IRE1 and PERK during lytic infection, strongly hinting at viral control of the UPR. We anticipate that these studies will help identify host factors required for productive infection, that could provide unique opportunities for the development of targeted therapeutics.
Researchers

Catya Faeldonea
Catya is a MA student investigating the single cell transcriptome of KSHV infected cells and the effect of modulating translation on lytic reactivation.

Guillermo Najarro
Guilermo is a Ph.D. student who is exploring the dynamics of the UPR in cells infected with KSHV and is specifically interested in identifying viral factors affecting this cellular response

Adriana Ramirez Negron
Adriana is exploring how KSHV modulates the UPR in a host cell and specifically, what role ATF6 and its targets play in KSHV infection.