How do cells sense, respond, and adapt to protein-folding stress or harmful RNAs?
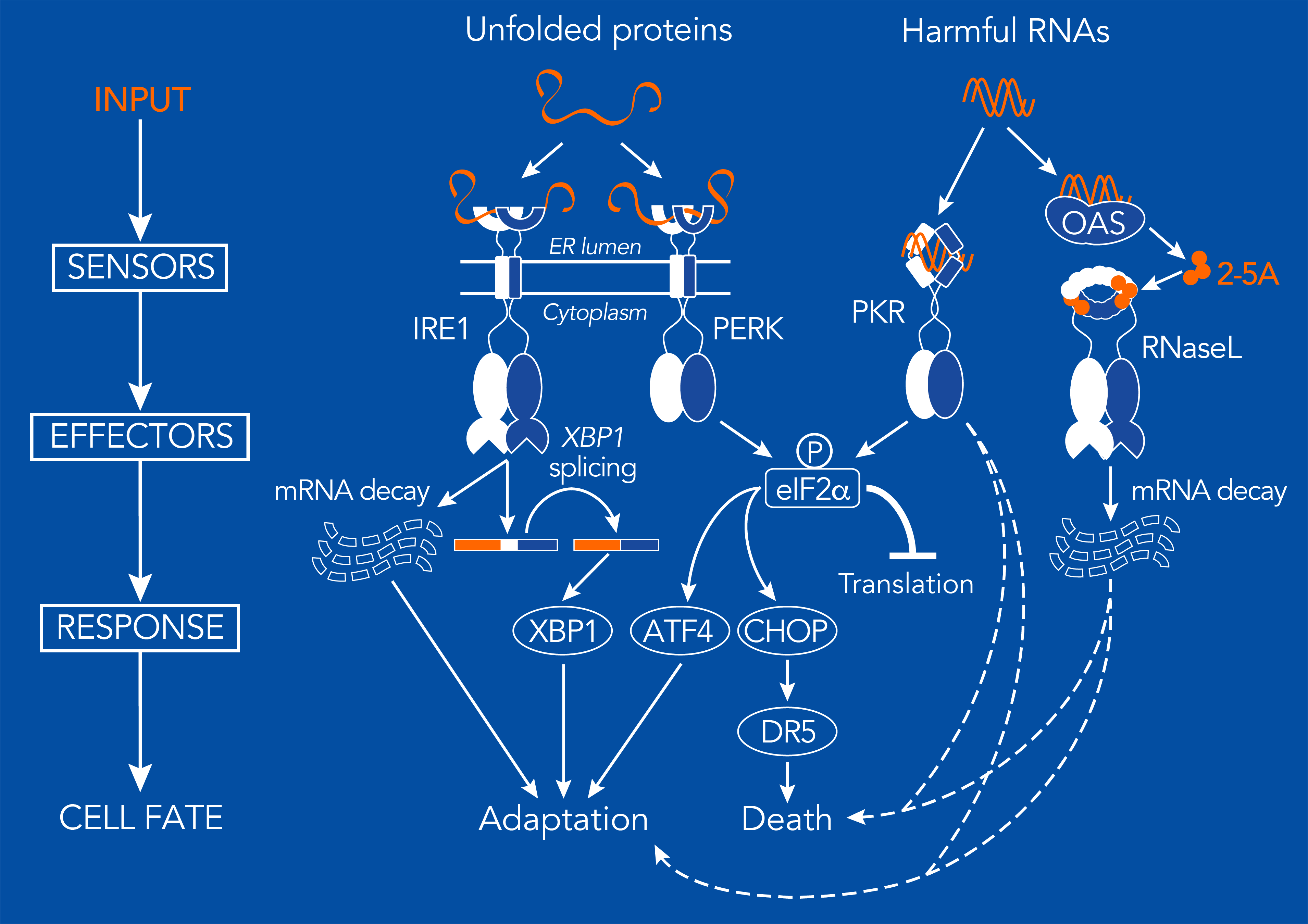
Maintaining compositional equilibrium -homeostasis- is essential for all biological systems. Cells employ complex mechanisms, collectively known as cellular stress responses, to detect internal imbalances and react to them. Deviations from homeostasis are quickly and effectively identified by specialized sensor proteins that relay information to downstream effectors, which initiate appropriate actions to restore homeostasis. When homeostasis cannot be restored, these mechanisms switch from pro-survival to pro-apoptotic roles, eliminating injured o diseased cells for the benefit of the organism. The Unfolded Protein Response (UPR), the stress response network of the endoplasmic reticulum (ER), exquisitely illustrates this concept. In the UPR, stress sensors recognize unfolded proteins in the ER lumen and relay this information to the nucleus. Comprehensive gene expression programs are subsequently initiated, allowing the cell to either resolve the protein folding stress or die trying. Cells detect and react to harmful RNAs in similar fashion. Unlike DNA, RNA molecules can adopt a broad range of structures, some of which are harmful to cells, for example, double-stranded RNA, which is often observed during viral infection. But harmful RNA structures can also occur in endogenous RNAs, and harmful self-RNAs elicit similar responses in the absence of viruses, supporting the notion of a fundamental homeostatic mechanism that maintains the health of the transcriptome. We refer to this central homeostatic mechanism as the “RNA stress response” (RSR). Moreover we hypothesize that signal integration in the UPR and in the RSR can help tailor exquisite cellular responses, promoting adaptation or clearing injured cells.
The modular nature of stress response signaling networks
The UPR and RSR are built upon signaling modules consisting of sensors and effectors. They both exert regulatory control by shutting down protein synthesis, turning on genes, and degrading transcripts. Specialized sensors recognize harmful macromolecules, either unfolded proteins in the UPR or potentially harmful RNAs in the RSR. In the UPR, the sensor proteins are the kinase PERK and the kinase/endoribonuclease IRE1 work together to recognize and respond to unfolded proteins. Activation of PERK elicits a transient global protein synthesis shutdown through the phosphorylation of the alpha subunit of the eukaryotic translation initiation factor 2 (eIF2α). This event leads to the paradoxical translation of a few select mRNAs that contain regulatory upstream open reading frames (uORFs), such as the mRNAs encoding the transcription factors ATF4 and CHOP. On the other hand, activation of IRE1 results in the production of the transcription factor XBP1 through a well-understood unconventional splicing mechanism. XBP1 turns on a vast gene expression program that alleviates ER stress. Active IRE1 also degrades ER-bound mRNAs, thereby lowering the load on the organelle.
In the RSR, similar systems operate to clear RNA stress. Harmful structured RNAs (e.g., double-stranded RNA) directly activate the kinase PKR and indirectly activate the pseudokinase/endoribonuclease RNaseL. Active PKR phosphorylates the same residue targeted by PERK on eIF2α, while active RNaseL degrades RNAs. Sensors and effectors are coupled as specific domains of single proteins in PERK, PKR and IRE1. RNaseL does not sense toxic RNAs directly. Instead RNaseL is activated by 2’,5’-linked oligoadenylates (2-5As), small RNA-like molecules generated by 2’,5’-oligoadenylate synthetases (OASs), which work as direct sensors of double-stranded RNA.
By virtue of being homeostatic mechanisms, and regardless of the input signal, both the UPR and RSR control cell fate. In collaboration with the group of Avi Ashkenazi (Genentech-UCSF), we have discovered a unifying mechanism that controls cell fate in cells undergoing ER stress (Lu et al., Science 2014). Here, a tug of war between pro-survival signals downstream of IRE1 and pro-apoptotic signals downstream of PERK determines whether a cell lives or dies. PERK activation results in expression of CHOP, which turns on the gene encoding the death receptor 5 (DR5), whose ligand-independent activation pushes the cell towards death. Active IRE1 combats the apoptotic signal by degrading the DR5 mRNA. Despite their high degree of mechanistic similarity and interconnectivity, we still do not know in detail how both stress responses may talk to each other in different stress instances. Since PERK and PKR converge on eIF2α, and because of the exceptional architectural similarities between both networks, we anticipate that mechanistic parallels between the UPR and RSR could explain how cells respond and adapt-or commit to apoptosis-upon encountering RNA stress.
Towards a network-level understanding of signal integration in cellular stress responses
The signaling mechanisms governed by the UPR have been extensively studied for the past two decades. However, our knowledge of the interactions between the UPR and other stress signaling pathways is limited. We hypothesize that the concerted actions of multiple signaling stress signaling pathways drive the homeostasis versus death decision of cells, and thus, we anticipate that understanding the degree of interconnectivity and signal integration among these pathways would be a major advancement in stress biology. With regards to the RSR, our understanding is even more limited. For example, while the roles of PKR and OAS-RNaseL are well understood in viral infections, we lack an in-depth understanding of how the pathways governed by these molecules operate in the absence of viruses. Signaling initiated by PKR and RNaseL have been implicated in a myriad of biological processes, including cognition, cell migration, cell adhesion, metabolic stress, and cancer. Thus, gaining a detailed, in-depth understanding of the RSR has profound biological and biomedical repercussions. Furthermore, we do not know the full complement of nodes and pathways that make up the RSR, nor do we know how, when and if they cross-talk to other stress signaling pathways, like the UPR. Moreover, our knowledge of the identities of endogenous RNAs that may be perceived as stress signals by the cell in different contexts remains limited to a few examples. To gain network-level insights on how stress responses cross-talk and how the RSR operates, we employ cutting-edge methods to determine the defining features of harmful RNAs, high-throughput techniques to identify harmful RNAs and RNA-protein interactions in living cells, and CRISPR-based genetic screens to identify genetic interactions between the genes that control the UPR and the RSR and the rest of the genome. Coupling these methods to cell biology and biochemistry allows us to deploy a comprehensive toolbox to mechanistically dissect the inner workings of stress response signaling in cells, and to understand whether this network is remodeled in disease. Follow the links “Harmful RNAs as stress signals”, “Genetic modulators of stress responses”, and "Network re-wiring in disease" to learn more about the approaches we take to answer these fundamental questions.



